 Encyclopædia Neurochirurgica
Encyclopædia Neurochirurgica
 Epilepsie associée aux gliomes infiltrants de l’adulte
Epilepsie associée aux gliomes infiltrants de l’adulte


V. Principes physiopathologies du processus pathologique
V.1. Approche tumorocentrique de l’épileptogenèse
V.1.1. Contraintes spatiales
L’effet de masse lié au développement du gliome a été proposé comme hypothèse dès les années 1940 par W. Penfield, neurochirurgien à Montréal, du fait de l’observation de modifications électrophysiologiques enregistrées en per-opératoire lors de l’ouverture durale après la levée du volet osseux (44, 63). L’effet de masse, lié à la prolifération tumorale, à l’œdème et à l’hypertension intracrânienne pourrait agir en altérant la microcirculation et favorisant des micro-ischémies péri-tumorales (5, 15, 63, 90, 101, 125, 131). Il est supposé que les gliomes infiltrants de bas grade sont plus volontiers épileptogènes par le biais d’une déafférentation progressive que les gliomes malins agiraient par le biais de dommages corticaux plus soudains comme l’ischémie, la nécrose ou l’hémorragie (9, 15, 101, 111, 125). Il n’existe cependant pas de corrélation claire entre le volume tumoral, le degré d’effet de masse, la présence d’œdème, la présence de nécrose et le risque épileptique sur de larges séries (5, 63, 88, 125). Enfin, l’existence d’un lien entre la localisation anatomique de la lésion et le risque épileptique laissent penser que des interactions particulières entre la tumeur et le cortex, plutôt que les caractéristiques tumorales intrinsèques semblent impliquées dans l’épileptogenèse.
V.1.2. Déafférentation
Des réorganisations profondes des réseaux neuronaux sont mises en évidence en périphérie des gliomes infiltrants, à type de modifications cellulaires (pertes neuronales, pertes gliales, gliose astrocytaire, activation microgliale) et de réorganisations synaptiques (diminution des circuits inhibiteurs, renforcement des circuits excitateurs) (5, 32, 63, 111, 128). La magnétoencéphalographie démontre que les gliomes infiltrants induisent des changements de l’architecture des réseaux et de la connectivité fonctionnelle cérébrale (4, 14, 15). Ces altérations de connectivité fonctionnelle participent à la création de réseaux aberrants ayant une susceptibilité accrue pour l’épilepsie et de mécanismes compensatoires de synchronisation excessive (4, 14, 15, 32).
Ladéafférentation des neurones du cortex péri-tumoral entraine une altération de leurs propriétés électrophysiologiques (71, 72, 117, 125). Une remarquable étude micro-anatomique réalisée dans le néocortex siège d’une infiltration tumorale gliale a mis en évidence des anomalies en lien avec l’infiltration tumorale (71, 72) : 1) une gliose réactive marquée ; 2) un écartement des fibres myelinisées ; 3) une perte des axones myelinisés dans les zones les plus infiltrées ; 4) une satellitose péri-neuronale par des cellules gliales tumorales ; 5) une infiltration par de la microglie activée et par des astrocytes réactifs ; 6) une diminution des interneurones GABAergiques en lien avec l’infiltration tumorale ; 7) une diminution de l’expression de la glutamate décarboxylase en lien avec l’infiltration tumorale ; 8) la persistance d’une architecture normale des cellules pyramidales ; 9) une diminution des synapses GABAergiques sur les cellules pyramidales ; 10) un pattern d’expression redevenu normal dans les prélèvements péri-tumoraux sans infiltration tumorale. Les cellules pyramidales semblent donc survivent à l’infiltration tumorales mais apparaissent déconnectées d’un point de vue synaptique. A l’inverse, la perte neuronale semble plus marquée sur les interneuronesGABAergiques. L’ensemble de ces modifications peut concourir à affaiblir l’inhibition GABAergique et à renforcer l’excitabilité des cellules pyramidales dans le cortex péri-tumoral (71, 72). Des études histopathologiques de prélèvements opératoires de néocortex péri-tumoral siège d’une activité épileptiques ont ainsi confirmé une diminution significative de l’immunoréactivité à la somatostatine et au GABA sans modification structurelle des interneurones GABAergiques persistants (49, 119). Des enregistrements électrophysiologiques intracellulairesex vivode cellules pyramidales situées dans le néocortex péri-tumoral ont montré des réponses anormales dans 80% des cas avec perte de l’inhibition fonctionnelle (51, 63, 119, 120). Ces cellules pyramidales déchargeant par bouffées peuvent recruter une plus large population de neurones et favoriser ainsi la formation d’un réseau local cohérent et coordonné, facilitant l’épileptogénicité dans le néocortex péri-tumoral (77).
V.1.3. Activation microgliale et inflammation
Les cellules tumorales gliales exploitent leur environnement en détournant la fonctionnalité des cellules non tumorales (astrocytes, microglies, macrophages, lymphocytes, cellules stromales) pour gagner en ressources et faciliter leur progression (21,67). A ce titre, les gliomes infiltrants constituent une véritable maladie neuro-dégénérative. Les cellules microgliales, ou macrophages « résidants » du cerveau, sont d’un intérêt tout particulier dans l’épilepsie associée aux gliomes infiltrants (9, 67), la microglie pouvant constituer jusqu’à 30% de la masse tumorale des gliomes malins (21, 23, 67, 129) et son degré d’expression est corrélé au grade de malignité (129). La microglie est activée par les cellules tumorales gliales de leur voisinage via desfacteurs chémoattractants émis de façon paracrine (facteurs de croissance, chémokines, cytokines, protéines de la matrice extracellulaire) (21, 23, 67, 129). L’accumulation de glutamate extracellulaire dans la périphérie tumorale peut également conduire àune activation microgliale (81). Les facteurs sécrétés par les cellules tumorales gliales sont donc capables d’induire une activation microgliale qui participera à créer un environnement favorable au développement du gliome et de réprimer le phénotype pro-inflammatoire de la microglie (67, 129).
La microglie activée par les cellules gliales tumorales est capable en retour de favoriser la migration des cellules tumorales gliales par le biais de la production de métalloprotéinases et de favoriser leur prolifération par la production de facteurs de croissance et l’activation de la voie EGF/Pi3K/Akt (21, 23, 67). D’autre part, la microglie activée constitue une source de relargage anormal de glutamate dans l’espace extracellulaire car elle exprime le complexe Xc- qui relargue du glutamate et perd sa capacité à le recapter pour tamponner l’espace extra-cellulaire (16, 62). Il est ainsi probable que la microglie activée dans le cortex péri-tumoral ne participe pas à tamponner l’excès de glutamate extracellulaire mais, qu’à l’inverse, elle contribue à l’augmentation de la concentration extracellulaire en glutamate, participant à l’épileptogenèse (16).
V.1.4. Altérations vasculaires
Une altération de la barrière hémato-encéphalique peut s’observer au cours de l’évolution des gliomes infiltrants, notamment lors de la transformation maligne mais également au décours de crises d’épilepsie (97, 98). Ces altérations de la barrière hémato-encéphalique sont épileptogènes : des crises d’épilepsie ont été observées chez 25% des patients chez qui la barrière hémato-encéphalique a été ouverte transitoirement par choc osmotique pour favoriser la pénétration intra-cérébrale de chimiothérapies (70). La rupture de la barrière hémato-encéphalique expose ainsi le tissu cérébral à desconcentrations fortes et prolongées en composants habituellement confinés au secteur plasmatique (albumine, fibrinogène, glutamate, immunoglobulines facteurs de croissance). Le glutamate relargué participe à l’augmentation de sa concentration extracellulaire. L’albumine relarguée est captée par les astrocytes environnants ce qui induit une down-régulation de l’expression de Kir4.1 dans ces astrocytes, avec pour conséquence une diminution des capacités de recapture de K+, l’ensemble entraînant une augmentation du K+ extracellulaire (58). De plus, l’albumine et le fibrinogène peuvent favoriser la mise en place d’une gliose astrocytaire réactive (16, 30) pouvant entrainer des troubles de l’homéostasie extracellulaire en glutamate et K+. Enfin, la fuite d’immunoglobulines induit une captation sélective par les neurones environnants, entraînant une dégradation neuronale progressive débutant sur les interneurones GABAergiques (76, 98). L’ensemble de ces modifications concourt à l’hyperexcitabilité des cellules pyramidales.
V.1.5. Facteurs génétiques
Aucun gène de susceptibilité associé à l’épilepsie tumorale n’a encore clairement été identifié. Une étude rapporte que, dans une population de 103 gliomes infiltrants de bas grade, l’absence de délétion 19q est associée à un risque accru de crise d’épilepsie et, particulièrement, de crises secondairement généralisées (52). Il est ainsi proposé que les gènes candidats de l’épilepsie tumorale pourraient se situer en 19q (52, 103). Il n’a pas été mis en évidence de lien entre le statut de co-délétion 1p19q et le risque épileptique (52, 78). La présence d’une mutation IDH1/2 est associée à un risque accru d’épilepsie dans les gliomes infiltrants de bas grade (68, 118, 136). Le gène suppresseur de tumeur LGI1 est impliqué dans la progression des gliomes et il n’est pas exprimé dans les gliomes malins (125). Il est également associé à une épilepsie à transmission autosomique dominante (45), mais aussi à des syndromes acquis dysimmunitaires impliquant la protéine LGI1 et certains auteurs suggèrent sa participation dans l’épilepsie tumorale (15, 32, 125). Dans une série de 77 glioblastomes, il a été récemment mis en évidence un lien entre une expression basse des gènes OLIG2 (Oligodendrocyte lineage transcription factor 2) et RTN1 (Reticulon 1) chez les patients présentant une épilepsie (66).
V.1.6. Equilibre acido-basique
Les gliomes présentent une hétérogénéité métabolique avec notamment une différence métabolique entre le centre tumoral et la périphérie lésionnelle (83). Cettehétérogénéité est retrouvée en spectroscopie IRM par résonance du phosphore, qui montre que le pH intracellulaire est légèrement basique dans le compartiment tumoral par rapport à la périphérie du gliome et au cortex contrôle (41). L’observation d’un pH alcalin dans les gliomes est à relier à l’augmentation de la concentration en cellules tumorales gliales qui sont plus alcalines que les neurones et que les cellules gliales non tumorales (5, 75). Le stress oxydatif est également à prendre en compte pour interpréter les variations de pH. Les gliomes malins présentent une diminution des médiateurs du stress oxydatif, une augmentation des enzymes glycolytiques et une production énergétique prédominante par glycolyse et acidose lactique, y compris en conditionsnormoxiques (111, 125). Deux études de microdialyse menées sur des gliomes malins montrent que la portion tumorale présente, par rapport au tissu péri-tumoral, une diminution de glucose, une augmentation des lactates, du glutamate et du glycérol (73, 102). Ainsi, il est retrouvé au sein du gliome un pH alcalin en cas de normoxie avec ATP haut mais un pH acide en cas d’hypoxie avec ATP bas lié à l’altération de l’apport vasculaire et à l’anaérobiose (83). A l’inverse, il est retrouvé au sein de l’œdème péri-lésionnel un pH alcalin lié à la normoxie du tissu péri-lésionnel qui est métaboliquement intègre et une diminution de l’ATP liée à l’augmentation du secteur extracellulaire d’origine vasogénique (83).
Ces modifications de l’équilibre acido-basique peuvent être épileptogènes. L’alcalinisation a des vertus épileptogènes par le blocage des conductances potassiques entrantes, l’inactivation des canaux sodiques et des conductances calciques dépendantes du K+ et via l’ouverture de jonctions gap. De plus, l’alcalinisation peut avoir des effets inhibiteurs sur les conductances médiées par le GABA (108). L’acidification peut causer des dégâts aux cellules gliales avoisinantes et, en condition d’acidose extracellulaire, les astrocytes sont sujets à une vulnérabilité par privation en oxygène et glucose (43, 111).
V.1.7. Jonctions inter-cellulaires
Les altérations de l’expression des connexines, qui sont observées dans les gliomes infiltrants, peuvent favoriser l’épileptogenèse (5, 32) : 1) l’augmentation des connexines peut favoriser la propagation de vagues calciques et contribuer à l’hypersynchronisation locale (32, 84, 111) ; 2) la perte de connexines fonctionnelles peut réduire les capacités de drainages hydro-électrolytiques et réduire la transmission des signaux inhibiteurs (3, 63, 115). A l’inverse, certains auteurs proposent que l’altération de l’expression des connexines, qui augmente avec le grade de malignité, pourrait avoir un effet protecteur vis à vis de l’épilepsie, expliquant ainsi le plus bas risque épileptique dans les gliomes malins (3, 111, 115).
V.1.8. Spiking glioma cells
La théorie des spiking glioma cells est née de deux études expérimentales électrophysiologiques qui ont suggéré que les cellules tumorales gliales étaient capables de générerdes potentiels d’action du fait d’une expression forte de canaux sodiques voltage-dépendants à leur surface (65, 93). Cependant, une étude complémentaire n’a pas mis en évidence de cellules gliales tumorales présentant des canaux sodiques voltage-dépendants aux sites des activités épileptiques (74, 94). De plus, une troisième étude a montré que les cellules tumorales étaient suffisamment dépolarisées pour générer des potentiels d’action via une augmentation des courants sodiques dans les conditions expérimentales (13). Il est peut probable que de tels « spikes gliaux » puissent survenir in vivo car, au potentiel de repos des cellules tumorales gliales, les canaux sodiques sont pleinement inactivés. Et de conclure qu’il s’agit d’une construction expérimentale : « a biophysical exercice with probably little physiological significance » (13).
V.1.9. Neurogenèse
La neurogenèse, aboutissant à des neurones néoformés ayant des caractéristiques immatures, pourraient rendre compte de neurones dépolarisés par le GABA etfavorisant l’épileptogenèse. Une neurogenèse a déjà été mise en évidence dans le tissu épileptique non tumoral (7), favorisée par les crises (92). Dans le tissu humain, des progéniteurs neuraux ont été identifiés chez l’adulte (106). De plus, les progéniteurs neuraux sous-ventriculaires peuvent être mis en jeu dans les mécanismes de réparation de lésions corticales (105). Il n’a cependant pas jusque là été mis en évidence de neurogenèse dans le cortex péri-tumoral des gliomes infiltrants.
V.1.10. Autres altérations possiblement épileptogènes
Des modifications enzymatiques sont observées dans les gliomes infiltrants, impliquant la glutamine synthétase, la glutamate décarboxylase, les glutaminases (121). Elles peuvent rendre compte d’une altération de lasynthèse des neurotransmetteurs, de leur stockage, de leur relargage, de leur recapture et de leur dégradation (32, 108). Une modification du métabolisme lipidique peut altérer l’équilibre membranaire et précipiter les dégâts cellulaires (108). Du fer peutêtre libéré dans l’espace extracellulaire, dans et à la périphérie d’une tumeur, suite à des saignements à partir des vaisseaux néo-formés (5, 9, 47, 113, 114). Le fer induit une peroxydation des membranes plasmiques et peut contribuer à la genèse d’activités épileptiques. Le relargage d’adénosine diminue significativement les réponses dépolarisantes au GABA, réduit la libération de glutamate au niveau pré-synaptique via les récepteurs A1 et module l’activité de récepteurs post-synaptiques (17, 24, 55). Une modification du Mg2+ extracellulaire a été rapportée (5).
V.2. Approche épileptocentrique de l’épileptogenèse, rôles du glutamate
V.2.1. Glutamate en conditions physiologiques
En conditions physiologiques, après avoir été libéré dans l’espace synaptique, le glutamate est transporté dans l’espace pré-synaptique neuronal ou capté par les astrocytes environnants et la microglie (31). Les concentrations extracellulaires en glutamate sont étroitement régulées par des transporteurs membranaires astrocytairesdépendant du Na+ (excitatory amino acid transport, EAAT 1 à 4) (31, 81, 128, 135). Les astrocytes sont très majoritairement responsables de la recapture du glutamate depuis la fente synaptique, la participation de la microglie est moindre (38, 40, 46, 48,62, 113, 115, 135). Cette clairance rapide du glutamate médiée par les EAAT est importante pour maintenir des concentrations extracellulaires basses en glutamate, n’excédant pas 1 à 3 μM, nécessaires pour assurer une transmission synaptique normale et éviter l’excitotoxicité induite par le glutamate (16, 31, 128). En parallèle, le glutamate peut également être relargué par les astrocytes par différents mécanismes (libération vésiculaire, action réverse des EAAT, hémi-canaux) jouant alors le rôle d’un glio-transmetteur, qui vient moduler la synchronisation des cellules pyramidales avoisinantes dans un périmètre de 100 μm (1, 2, 31).
V.2.2. Altération de l’homéostasie du glutamate dans les cellules gliales tumorales
Des dysfonctions du transport du glutamatesont identifiées dans les gliomes infiltrants et les cellules gliales tumorales agissent à l’opposé des astrocytes non tumoraux en relarguant du glutamate au lieu de le capter (31, 133).
La recapture du glutamate est altérée dans les cellules gliales tumorales qui perdent la fonctionnalité des transporteurs du glutamate EAAT (31, 107, 130). De plus, les cellules gliales tumorales larguent du glutamate. Il en résulte un excès de glutamate extracellulaire dans l’environnement des gliomes infiltrants, mesuréentre 100 et 500 μm au sein du gliome et dans sa périphérie (6, 73, 102, 122, 128). Chez l’homme, une étude incluant 288 gliomes infiltrants, a retrouvé des taux plus important de glutamate et une altération de l’expression des transporteurs du glutamate dans la tumeur et dans le cortex péri-tumoral chez les patients présentant une épilepsie par rapport à ceux n’en présentant pas (135). L’augmentation de glutamate dans la périphérie des gliomes en comparaison avec le cerveau non envahi a également été miseen évidence in vivo par spectroscopie IRM (99). Les cellules tumorales gliales libèrent du glutamate par une action différente des astrocytes non tumoraux via le complexe d’échange cystine/glutamate (Xc-), un transporteur couplé cystine-glutamate qui libère du glutamate dans l’espace extracellulaire en échange d’une captation intracellulaire de cystine (16, 31, 107). La principale fonction de ce système Xc- est la captation de cystine qui servira à la synthèse de glutathion, un agent antioxydant, afin de protéger la cellule tumorale gliale contre les effets des radicaux libres (12, 22, 31, 128). Le système Xc-, qui est à la fois une nécessité pour la survie des cellules tumorales gliales et une opportunité pour leur croissance tumorale (12, 22, 128).
Il semble exister une expression anormale de récepteurs ionotropiques et métabotropiques au glutamate dans les cellules tumorales gliales (32) : 1) une augmentation de l’expression des récepteurs métabotropiques au glutamate est observée dans le cortex péri-tumoral dont l’activation peut potentialiser l’excitotoxicité glutamatergique médiée par NMDA (32, 130, 132) ; 2) une modification de l’expression des récepteurs AMPA, qui prédominent au front de migration et aux sites d’adhésions entre cellule et matrice extracellulaire, peut favoriser la migration des cellules tumorales gliales (31, 69, 122).
V.2.3. Altérations de l’homéostasie du glutamate dans les cellules non tumorales
En parallèle au défaut de recapture du glutamate et à son relargage excessif par les cellules tumorales gliales, vient de surajouter une modification des capacités de tamponnage du glutamate par les astrocytes non tumoraux, la microglie et les neurones. Alors qu’ils sont censés capter l’excès de glutamate extracellulaire pour maintenir l’homéostasie, leurs capacités semblent dépassées dans les gliomes infiltrants (17, 31). En effet, la fonctionnalité EAAT est compromise dans les gliomes infiltrants par altération du gradient électrochimique pour le Na+ et le K+ qui est la principale force directrice de l’influx entrant de glutamate dans les astrocytes via EAAT (16, 122). Une diminution de l’activité de la glutamine synthétase est observée dans les astrocytes réactifs (81, 86). De plus, la microglie activée exprime le complexe Xc- et perd la fonctionnalité EAAT, elle peut ainsi relarguer du glutamate alors que sa capacité à le recapter est diminuée (16). Enfin, une baisse d’expression de EAAT est également observée dans les neurones (81).
V.3. Conséquences de l’augmentation du glutamate extracellulaire dans les gliomes infiltrants
La concentration extracellulaire en glutamate est fortement augmentée au sein et en périphérie des gliomes infiltrants du fait de : 1) un relargage massif par les cellules tumorales gliales ; 2) un relargage par la microglie activée ; 3) une diffusion depuis la circulation systémique via des altérations de la barrière hémato-encéphalique ; 4) une diminution de la recapture par les cellules tumorales gliales, par la microglie activée, par les astrocytes réactifs non tumoraux et par les neurones. Il en résulte un excès majeur et stable de glutamate extracellulaire dans l’environnement des gliomes infiltrants qui est responsable de trois effets majeurs associés au développement tumoral : 1) neurotoxicité ; 2) pro-invasif ; 3) pro-épileptique (12, 128).
V.3.1. Neurotoxicité induite au glutamate
La boite crânienne étant un milieu restreint, les gliomes infiltrants obtiennent de l’espace en détruisant de façon active les cellules avoisinantes. L’excès de glutamate extracellulaire dans l’environnement des gliomes infiltrants induit une hyperactivation neuronale prolongée médiée par les récepteurs NMDA, l’excitation induite devenant délétère et entrainant une mort neuronale (12, 16, 27, 107, 122). Les oligodendrocytes tolèrent mal desconcentrations extracellulaires en glutamate élevées et prolongées et meurent également (27). La capacité des astrocytes à résister à l’ischémie est d’ailleurs réduite par la perte fonctionnelle de EAAT même si les cellules gliales tumorales sont plutôt résistantes à l’excitotoxicité induite par le glutamate (130).
V.3.2. Glutamate, un facteur de croissance pour les gliomes
Le glutamate, par le biais de la voie des Pi3-kinases, agit comme un facteur de croissance pour les cellules tumorales gliales et leurs précurseurs (81, 109). La majorité des cellules tumorales gliales possède des récepteurs au glutamate à leur surface membranaire, y compris les brain tumor-initiating tumor cells (81, 82, 122). Le glutamate extracellulaire agit comme signal autocrine etparacrine pour promouvoir la migration et la prolifération des cellules tumorales gliales (12, 31, 69, 122, 128). L’activation des récepteurs ionotropiques AMPA des cellules gliales tumorales permet leur migration (augmentation de l’adhésion à la matriceextracellulaire, interactions entre matrice extracellulaire et intégrines, activation des kinases d’adhésion focale, détachement cellulaire) (31, 33). L’activation des récepteurs ionotropiques AMPA et des récepteurs métabotropiques va également favoriser la prolifération tumorale par une augmentation de l’expression du récepteur à l’EGF et activation de la voie des Pi3-kinases (12, 69, 128, 130).
V.3.3 Excitabilité neuronale induite par le glutamate
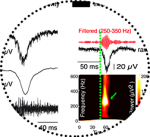
En se liant aux récepteurs des neurones pyramidaux dunéocortex péri-tumoral, le glutamate relâché par les cellules tumorales gliales contribue aux crises d’épilepsie (12, 128). L’augmentation des taux de glutamate extracellulaire est ainsi un facteur pronostic indépendant du risque épileptique chez ces patients (135). Une étude récente a étudié les liens entre l’augmentation du glutamate extracellulaire et la genèse des activités épileptiques dans un modèle de xénogreffes de gliomes humains chez la souris immuno-déficiente (17). Il a été mis en évidence parstéréoélectroencéphalographie chez l’animal vigil une activité épileptiforme dans le cortex péri-tumoral dans 37,2% des cas, la fréquence variant avec la progression tumorale. L’analyse électrophysiologique sur tranches a montré une augmentation du relargage de glutamate extracellulaire médiée par le système Xc- avec, pour résultante, une augmentation de l’excitabilité des cellules pyramidales dans le cortex péri-tumoral (Figure 2). En parallèle, il a été mis en évidence une diminution de l’inhibition GABAergique. Cette étude a permis de conclure que la diminution du seuil d’excitabilité des cellules pyramidales du cortex péri-tumoral pouvait être attribuée formellement à une augmentation de la signalisation glutamatergique (17).
Une étude électrophysiologique in vitro récente a mené des enregistrements du cortex péri-tumoral issu de patients opérés d’un gliome infiltrant (91). Des explorations pharmacologiques et des enregistrements intracellulaires ont permis d’identifier, pour la première fois chez l’homme, les réseaux locaux à l’origine des décharges épileptiformes inter-critiques spontanées. Ces activités sont bloquées par les antagonistes des récepteurs AMPA au glutamate (mais pas NMDA), confirmant la mise en jeu de la signalisation glutamatergique dansl’épileptogenèse des gliomes infiltrants (Figure 2) (91). Les enregistrements intracellulaires ont permis de préciser la séquence d’activation neuronale : les interneurones déchargeaient avant et durant la phase initiale de l’activité des neurones pyramidaux, suggérant un effet déclenchant (91).
V.4. Approche épileptocentrique de l’épileptogenèse, rôles du GABA
S’il est tentant de réduire l’implication GABAergique dans l’épilepsie à une simple diminution de l’inhibition GABAergique, les constats montrentqu’elle est beaucoup plus complexe dans l’épilepsie associée aux gliomes infiltrants : diminution de l’inhibition, excitabilité paradoxale, modification de l’expression des récepteurs (111).
V.4.1. Altérations de la signalisation GABAergique inhibitrice
Une diminution de l’inhibition GABAergique est observée, touchant à la fois la concentration en GABA, l’expression des récepteurs au GABA et leur fonctionnalité, la perte en neurones GABAergiques avec appauvrissement de leur arborisation et, à plus large échelle, une altération des réseaux inhibiteurs GABAergiques. Elle peut participer à l’épileptogenèse en induisant une diminution de l’inhibition nécorticale dans le cortex péri-tumoral (56).
L’infiltration tumorale induit une déafférentation des neurones pyramidaux dans le néocortex péri-tumoral avec diminution des synapses GABAergiques sur les cellules pyramidales et une diminution des interneurones GABAergiques (71, 72, 132). Une étude par microdialyse menée sur des gliomes malins retrouve une augmentationde la concentration en GABA dans le tissu péri-tumoral, inférieure à celle du centre de la tumeur, suggérant une perte de l’inhibition GABAergique dans la périphérie des gliomes (11). L’analyse électrophysiologique sur tranches dans un modèle murin de xénogreffes de gliomes humains a montré, en parallèle de l’augmentation du relargage de glutamate extracellulaire, une diminution de l’inhibition GABAergique (17). Ces données ont d’ailleurs été confortées par des enregistrements électrophysiologiques intracellulaires in vitro de cellules pyramidales situées dans le néocortex péri-tumoral chez l’homme montrant une perte de l’inhibition fonctionnelle et un pattern de décharge en bouffées (51, 63, 119, 120). L’ensemble de ces modifications peut concourir à affaiblir l’inhibition GABAergique et à renforcer l’excitabilité des cellules pyramidales dans le cortex péri-tumoral. La perte de l’inhibition GABAergique est également montrée de façon indirecte par magnétoencéphalographie (20). Sur une série de 12 patients,dont 8 étaient porteurs d’un gliome infiltrant, il est montré un ratio P35m/N20m supérieur dans l’hémisphère lésionnel que dans l’hémisphère controlatéral chez les patients porteurs d’une épilepsie par rapport aux patients sans épilepsie. L’activité P35m étant liée à l’inhibition post-synaptique des cellules pyramidales de la couche granulaire, son augmentation suggère une diminution de l’inhibition GABAergique dans l’hémisphère lésionnel et pourrait participer à l’épileptogénicité (20).
Cependant, une étude électrophysiologique in vitro récente a mené des enregistrements du cortex péri-tumoral issu de patients opérés d’un gliome infiltrant (91). L’exploration pharmacologique et électrophysiologique des réseaux neuronaux l’origine des décharges épileptiformes spontanées à montré, pour la première fois chez l’homme, outre la participation de la signalisation glutamatergique AMPA, une participation de la signalisation GABAergique, les activités épileptiformes étant bloquées par l’administration d’antagonistes GABAa (91) (Figure 2). De plus, les enregistrements intracellulaires ont montré que les interneurones GABAergiques étaient les initiateurs de ces décharges et précédaient les décharges des neurones pyramidaux, suggérant un effet déclenchant (91). Ainsi, cette étude originale suggère que l’inhibition GABAergique n’est pas simplement diminuée dans le néocortex péri-tumoral des gliomes infiltrants mais que les modifications de la signalisation GABAergique sont plus profondes : elle semble détournée, passant d’une fonction inhibitrice à une fonction déclenchante, voire excitatrice.
V.4.2. Action anti-proliférante de la signalisation GABAergique
Les cellules tumorales gliales présentent des modifications de l’expression des récepteurs au GABA (32). Il apparaît que les gliomes expriment des récepteurs GABAa qui peuvent être fonctionnels et que cette expression diminue avec l’augmentation du grade de malignité (65, 112, 134). La fonctionnalité des récepteurs GABAa diminue la prolifération cellulaire, comme en témoigne la relation inverse entre leur expression et le grade de malignité (59, 112, 134). Le GABA étant impliqué dans la prolifération des neurones immatures et dans leur différentation, il pourrait participer à limiter la prolifération des gliomes. Ainsi, concernant la signalisation GABAergique des cellules gliales tumorales, il semble qu’elle agisse comme un frein sur la prolifération cellulaire, les cellules tumorales devant s’en affranchir pour gagner en capacités de prolifération. Le GABA agit ici comme un facteur anti-proliférant.
V.4.3. Troubles de l’homéostasie du chlore dans les cellules gliales tumorales
Une forte concentration intracellulaire en Cl- est nécessaire à la migration et à la prolifération des cellules tumorales gliales, justifiant son accumulation intracellulaire (113). Les cellules tumorales gliales maintiennent donc de fortes concentrationsintracellulaires en Cl- entre 80 à 140 mM (47, 113, 128). Ceci est permis par une modification de l’expression des co-transporteurs du Cl-. NKCC1, un co-transporteur permettant d’accumuler le Cl- en intracellulaire, constitue le moyen principal d’accumulation de Cl- dans les cellules tumorales gliales (37, 38, 40, 46). L’expression protéique de NKCC1 est corrélée au grade de malignité (40). Il est observé en parallèle une altération de l’expression de KCC2, un co-transporteur du Cl- permettant de le faire sortir de la cellule (38, 48, 113, 114). Cette accumulation intracellulaire de Cl- est nécessaire pour permettre les modifications de volume cellulaires durant la prolifération et la migration des cellules tumorales gliales (47).
V.4.4. Troubles de l’homéostasie du chlore dans les neurones du néocortex péri-tumoral
Des changements de la concentration intracellulaire neuronale en Cl- sont retrouvés dans le cortex péri-tumoral (10). Ce constat a une implication directe sur la signalisation GABAa. La conséquence de l’activation des récepteurs GABAa en terme de dépolarisation/hyperpolarisation dans un neurone dépend des facteurs qui contrôlent les gradients de Cl- et d’HCO3- qui peuvent les traverser. En conditions pathologiques, on peut assister à une « régression fonctionnelle » des récepteurs GABAa qui est très souvent associée à une diminution de la fonctionnalité KCC2, la fonctionnalité NKCC1 pouvant ne pas intervenir (39). Il s’opère une inversion de la fonctionnalité GABAergique de réponses hyperpolarisantes au GABA, sous-tendant l’inhibition classique, à des réponses dépolarisantes, qui peuvent devenir excitatrices. De nombreuses conditions pathologiques peuvent d’ailleurs induire des réponses dépolarisantes au GABA (trauma, température élevée, chocs osmotiques, transsection nerveuse, privation d’oxygène et de glucose) (25, 80, 104, 116, 123, 124). Des réponses dépolarisantes au GABA ont d’ailleurs été mises en évidence dans l’épilepsie mésio-temporale avec sclérose de l’hippocampe (53, 54).
Le développement d’un gliome infiltrant peut être considéré comme une agression cérébrale, et ce, non uniquement par la possible déafférentation induite par la croissance tumorale. L’exposition de neurones hippocampiques de souris à un milieu ambiant de gliomes provenantde cultures de lignées de glioblastomes induit l’augmentation rapide et transitoire de leur concentration intracellulaire en Cl- (10). L’augmentation intracellulaire du Cl- dans les neurones semble assurée par la participation des co-transporteurs du Cl-(10, 18). L’augmentation intracellulaire du Cl- dans les neurones est par ailleurs médiée par les récepteurs ionotropiques au glutamate NMDA et AMPA, comme dans d’autres conditions pathologiques. La libération de glutamate par les cellules tumorales gliales altère donc l’homéostasie du Cl- dans les neurones avoisinant (10). Une étude électrophysiologique in vitro à partir de l’inclusion de prélèvements membranaires de gliomes infiltrants dans des oocytes de grenouille a mis en évidence des courants médiés par le GABA ayant un potentiel d’inversion plus dépolarisé que celui du cortex contrôle en lien avec une altération de l’expression des co-transporteurs du Cl- (NKCC1 et KCC2) pouvant résulter en une diminution de l’inhibition GABAergique ainsi qu’en des effets dépolarisants (24). Une autre étude récente de co-cultures de neurones en présence de cellules tumorales gliales retrouve les mêmes anomalies (34). Ces préparations expérimentales sur un modèle animale suggèrent que le GABA est impliqué à la fois dansla croissance tumorale des gliomes infiltrants et dans l’épileptogenèse qui leur est associée (18). Une altération de la signalisation GABAergique est enregistrée ainsi qu’une expression anormale des co-transporteurs du Cl- et l’accumulation de Cl- intracellulaire pourrait rendre compte de réponses dépolarisantes au GABA contribuant à la genèse des activités épileptiques dans le cortex péri-tumoral. Ces données ont été reproduites chez l’homme par une étude électrophysiologique in vitro récente menée sur du cortex péri-tumoral issu de patients opérés d’un gliome infiltrant (91). Les explorations pharmacologiques et les enregistrements intracellulaires ont démontré que les décharges épileptiformes et qu’elles étaient initiées par les interneurones GABAergiques. En réponse à la décharge des interneurones, les neurones pyramidaux présentent des anomalies de réponse à la neurotransmission GABA qui ont un effet excitateur sur 65% des neurones pyramidaux, au lieu d’être inhibiteur. Ces résultats suggèrent que deseffets dépolarisants, et potentiellement excitateurs, de la signalisation GABAergique sont en cause. L’étude de la régulation du Cl- au sein des neurones pyramidaux du néocortex péri-tumoral a montré que le gradient électrochimique était en faveur de réponses dépolarisantes au GABA dans 60% des cas, témoignant d’une concentration intra-neuronale anormalement élevée en Cl-. Ces anomalies étaient de plus corrélées au degré d’infiltration du néocortex par des cellules tumorales (82% des fortes infiltrations et44,4% des faibles infiltrations ; 100% des glioblastomes, 57% des gliomes anaplasiques et 33,3% des gliomes infiltrants de bas grade). Les co-transporteurs du Cl- KCC2 et NKCC1 ont alors été explorés, montrant une expression de NKCC1 augmentée et une expression de KCC2 diminuée dans le néocortex péri-tumoral. Ces données confirment le rôle d’une perturbation de l’homéostasie du Cl-, avec une concentration neuronale anormalement élevée en Cl- par une altération de l’expression de ses co-transporteurs NKCC1et KCC2, rendant compte de réponses excitatrices au GABA dans la genèse des activités épileptiques associées aux gliomes infiltrants chez l’homme. Une étude expérimentale a étudié les mécanismes rendant compte d’une altération des co-transporteurs du Cl- (34). La présence de réponses dépolarisantes au GABA dans les neurones pyramidaux entourés de cellules tumorales gliales est en partie due à une réduction de la fonctionnalité de KCC2 médiée par une augmentation du zinc intracellulaire venant activer la voie des Src/TrkB tyrosine kinases (34). Il n’existe pas d’étude démontrant le mécanisme de l’augmentation de NKCC1 dans les neurones du néocortex péri-tumoral. Il est cependant démontré que l’augmentation de NKCC1 dans les cellules tumorales gliales est médiée par la voie des PI3 et des WNK kinases (40).
V.5. Troubles de l’homéostasie du Potassium
Une des propriétés fondamentales des astrocytes est le contrôle de l’homéostasie ionique, notamment du K+ extracellulaire, pour contrôler l’excitabilité neuronale(16). La recapture du K+ est assurée principalement par les Gap-jonctions et par les canaux Kir (16, 85). On observe une absence d’expression de canaux Kir4.1 fonctionnels dans les gliomes (50, 85, 137). Cette perte fonctionnelle de Kir4.1 dans les gliomes infiltrants explique pourquoi les capacités de recapture du K+ par les cellules tumorales gliales sont altérées (127). Une forte augmentation de K+ extracellulaire est également nécessaire pour la prolifération et la migration des cellules tumorales gliales (47, 64, 114, 128). L’homéostasie du K+ peut ainsi être altérée dans les gliomes infiltrants et l’augmentation de la concentration extracellulaire en K+ dans le cortex péri-tumoral peut rendre compte d’une augmentation de l’excitabilité des cellules pyramidales (26).
V.6. Troubles de l’homéostasie de l’eau
L’œdème interstitiel présent dans les gliomes infiltrants résulte de la mort cellulaire induite dans sa composante cytotoxique et des réactions inflammatoires dans sa composante vasogénique (83). Ilexiste un lien clair entre la présence d’œdème et l’expression du système Xc-, faisant suggérer la participation de l’excitotoxicité du glutamate dans la genèse de l’œdème (36, 107). La microglie activée et les astrocytes réactifs participent également àla mise en place de l’œdème tumoral (36, 129). L’aquaporine 4, qui est le transporteur trans-membranaire principal de l’efflux d’eau couplé au K+ et au Cl- lors des modifications de volumes induites par les déplacements cellulaires est exprimée par les cellules tumorales gliales (35, 57, 127). Les flux hydro-électrolytiques engendrés lors des déplacements des cellules tumorales gliales peuvent favoriser des troubles hydro-électrolytiques épileptogènes.